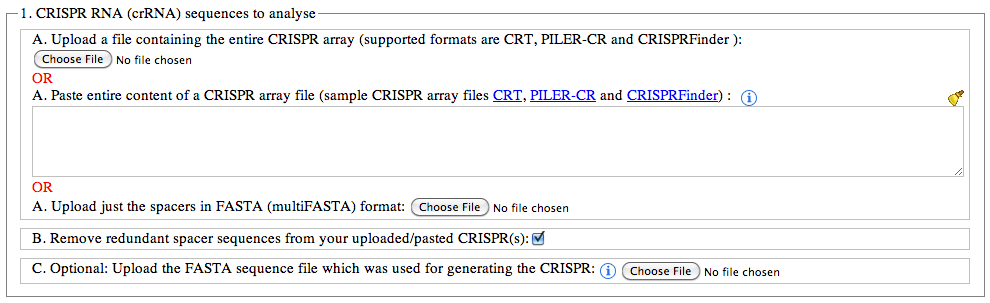
The format needs to be unedited as the program needs to extract specific information from the files. A multifasta file will work as long as the identifiers are unique. If the identifiers may not be unique- upload the fasta spacer file and a unique identifier will be added. For the CRISPRDetect format do not delete any lines. Missinglines e.g. for the flanking sequences might cause erors. If so add some sequence in.
An example CRISPRDetect file is here:
Allowed options:
-minNR minimum number of repeats a CRISPR must contain; default 3
-minRL minimum length of a CRISPR's repeated region; default 19
-maxRL maximum length of a CRISPR's repeated region; default 38
-minSL minimum length of a CRISPR's non-repeated region (or spacer region); default 19
-maxSL maximum length of a CRISPR's non-repeated region (or spacer region); default 48
-searchWL length of search window used to discover CRISPRs; (range: 6-9)
Allowed options: Basic options: -inSample PILER-CR fileSequence file to analyze (FASTA format). -out Report file name (plain text). -seq Save consensus sequences to this FASTA file. -trimseqs Eliminate similar seqs from -seq file. -noinfo Don't write help to report file. -quiet Don't write progress messages to stderr. Criteria for CRISPR detection, defaults in parentheses: -minarray Must be at least repeats in array (3). -mincons Minimum conservation (0.9). [At least N repeats must have identity >= F with the consensus sequence. Value is in range 0 .. 1.0. It is recommended to use a value < 1.0 because using 1.0 may suppress true arrays due to boundary misidentification.] -minrepeat Minimum repeat length (16). -maxrepeat Maximum repeat length (64). -minspacer Minimum spacer length (8). -maxspacer Maximum spacer length (64). -minrepeatratio Minimum repeat ratio (0.9). -minspacerratio Minimum spacer ratio (0.75). ['Ratios' are defined as minlength / maxlength, thus a value close to 1.0 requires lengths to be similar, 1.0 means identical lengths. Spacer lengths sometimes vary significantly, so the default ratio is smaller. As with -mincons, using 1.0 is not recommended.] Parameters for creating local alignments: -minhitlength Minimum alignment length (16). -minid Minimum identity (0.94).
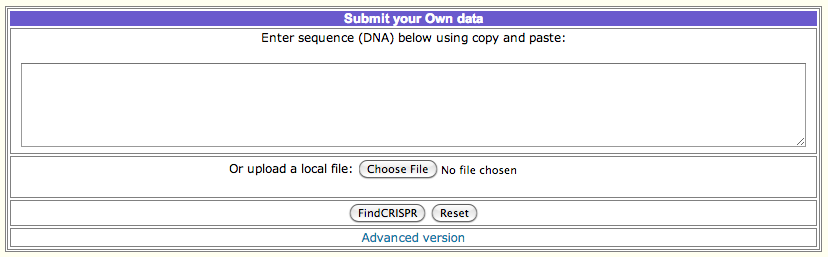
How to obtain CRISPRFinder output :
To perform a CRISPR prediction and obtaining the output file, follow the steps:
A. Upload or paste your genomic sequence in the corresponding text box and press submit.
B. The next page shows a table with headers 'Confirmed CRISPRs' and 'Questionable CRISPRs' along with links to the corresponding files.
Clicking on a link will take you to the corresponding CRISPR arrays visualization.
C. Click on the button named "Crispr Properties" will open the output file you need. Save the file, and you can use the file or its
content as input in CRISPRTarget.
[Note: You can concatenate all the predicted CRISPR Arrays (individual output files) in one file and upload/paste in CRISPRTarget.]
More detailed description can be found here.
Selected databases are provided in CRISPRTarget. The database updates are provided in the news.
In general Databases will be updated after each release of Genbank (even months) or RefSeq (odd months). The nt and env databases come from the BLAST databases and will be updated monthly.
Genbank (BLAST Nucleotide) databases, these are held locally but updated bimonthly with GenBank or RefSeq releases.
Databases are updated with full releases of Genbank and RefSeq (Note: Files (e.g. RefSeq plasmid.1.1.genomic.fna.gz. or GenBank gbphg1.seq.gz) files are downloaded, converted to fasta if needed, concatenated, converted to blast databases, and BLAST+ run locally)
CAMERA database: We included viral parts of the CAMERA databases. 913,9883 gene sequences, 1 Billion bases (Files: CAM_PROJ_ReclaimedWaterVirues.read.fa, CAM_PROJ_MarineVirome.read.fa, CAM_P_0000909.read.fa, CAM_P_0000792.read.fa). ACLAME. 125,190 sequences, 96 million bases (V0.04, 8/2009, last version).
IMG/VR database: IMG_VR: IMGVR_all_nucleotides.fna.gz . The current version is v4 Sept 2022 (or 6.1). IMG_VR_2022-09-20_6.1 - IMG/VR v4 - high-confidence genomes only (~80Gb). First version provided from 7/2018, legacy version in in the test directory 43 Gb Oct 2020 v3 or 5.1
This includes IMGVR viral contigs with IDS like IMGVR_UViG_2579779064_000006|2579779064|2579849396|1-17041, some genomes from RefSeq with IDs like NC_027986.1, and other entries Gammaproteobacteria_gi_553770258, and UGV-GENOME-3293712.
To interpret the results you will need the information file downloaded from here (requires JGI registration): IMG_VR: IMGVR_all_Sequence_information.tsv
HUVirDB Database downloaded from here : HuVirDB Assemblies opengut.ucsf.edu/HuVirDB-1.0.fasta.gz Cite Soto-Perez et al., 2019, Cell Host & Microbe 26, 325–335 https://doi.org/10.1016/j.chom.2019.08.008
The default values used by NCBI BLASTn for short sequences <30 bases (defaults for long sequences are in brackets) are: Gap open -5(-5) Gap extend -2(-2) Match +1(+1) Mismatch -3(-3) Word size 7(11) Expect (E): 1000 (10) Filter: No (Yes) Blastn-short (noticed 8/2018) now uses +2/-3 more similar to +1/-1 used here. A 30 base exact matrch is 60 for blastn-short and 30 for CRISPRTarget The initial CRISPRTarget defaults are the same except that a gap is penalised more highly (-10), the mismatch penalty is -1 and the E filter is 1. In addition, there is also no filter or masking for low complexity. BLAST calculates the scores over the length of the match, and only shows this match. For example, a spacer of 32 bases that matches to a target in 17 of 20 bases would score 20-3=17 and 20 bases would be output. The expected (E) values of the match will be more likely to pass the filter if smaller databases are used (e.g. the default phg and plasmid). Changing BLAST parameters: Please note that only certain combinations of parameters produce valid statistics (this others will not work). For +1, -1 an attempt to use some combinations might fail. See the following example for allowed paramters: $ blastn -db database -query myseq -gapopen 1 -gapextend 1 -reward 1 -penalty -1 BLAST engine error: Error: Gap existence and extension values 1 and 1 are not supported for substitution scores 1 and -1 3 and 2 are supported existence and extension values 2 and 2 are supported existence and extension values 1 and 2 are supported existence and extension values 0 and 2 are supported existence and extension values 4 and 1 are supported existence and extension values 3 and 1 are supported existence and extension values 2 and 1 are supported existence and extension values 4 and 2 are supported existence and extension values Any values more stringent than 4 and 2 are supported (e.g. 10, 2) Suggestion: Useful changes might be: a. Reducing the gap penalty to 4 or 5 if you have reason to believe that gaps are tolerated in your system. b. Increasing the E to 10 or 100 in the unlikely event you are not getting hits. c. Increasing the mismatch penalty to -3 screens out mismatches. 6. Set the DB size (effective database size).
This is optional. This should be the total size of the databases you search. BLAST calculates the E (Expect) value based on the size of the database searched. If one search against multiple databases is done the database need not be specified as BLAST does it internally. To compare the significance of matches in two or more consecutive searches of different databases, this value should be set as the sum of the two databases sizes (e.g. for 270 Mb + 80 Mb= 350 Mb enter '350000000').
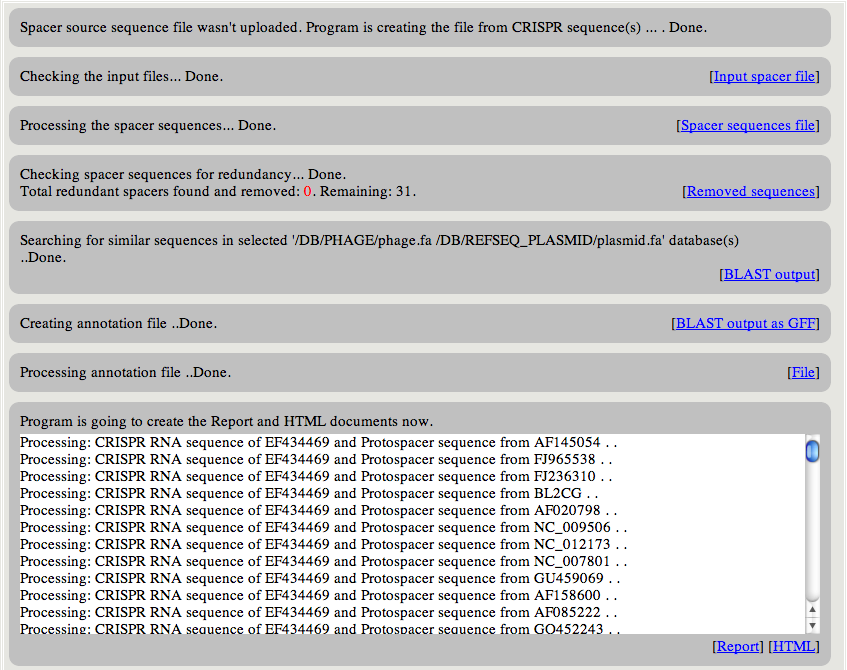
|
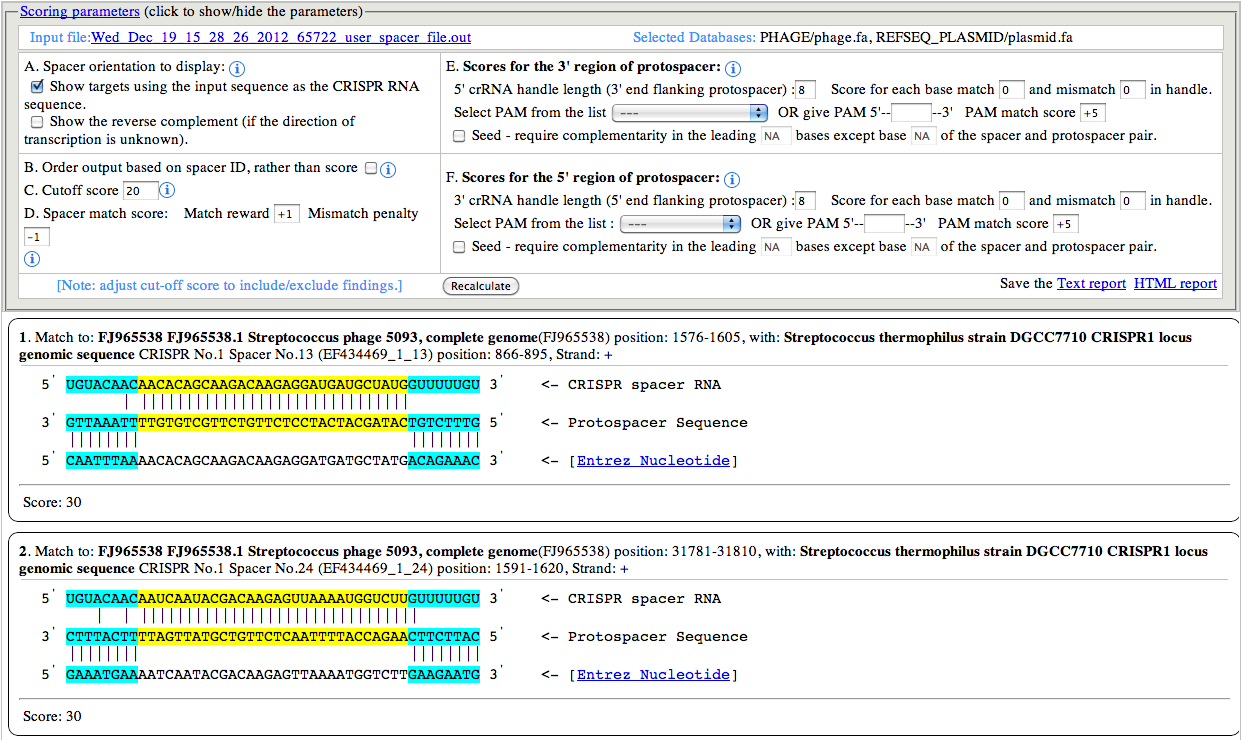
A. Spacer orientation to display:
By default the Spacer sequence (top most in any set) is shown in 5'
to 3' orientation, and the protospacer sequence (the target sequence
which base pairs with spacer sequence) is shown in 3' to 5'. However
user can choose to display the other strand of the Spacer sequence,
which brings the other strand of the protospacer sequence in the middle.
This option is especially useful when the orientation of the CRISPR
array is not known/certain.
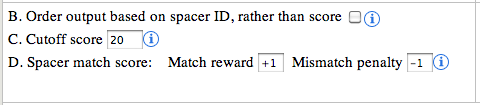
B. Order output based on spacer ID:
a spacer ID is represented with 3 elements, the sequence ID,CRISPR
Index and spacer Index, separated by underscore (e.g: EF434469_1_13). By
default the output is sorted in descending order of the calculated
score. However, if the user wants to show/arrange the output based on
the spacer ID, selecting this option will achieve that. This option can
be very useful in visually inspecting the output, as it maintains the
order of the spacer for every CRISPR.
C. Cutoff score:
The cutoff score is used for filtering out the low scoring matches
from the output. The default value is 20, but user can use any cutoff or
no cutoff value to show/hide matches.
D. Spacer match score:
The default values for match and mismatch are +1 and -1
respectively. These values along with the cutoff score provide a smart
way to push the matches with gaps down the order or even omit them from
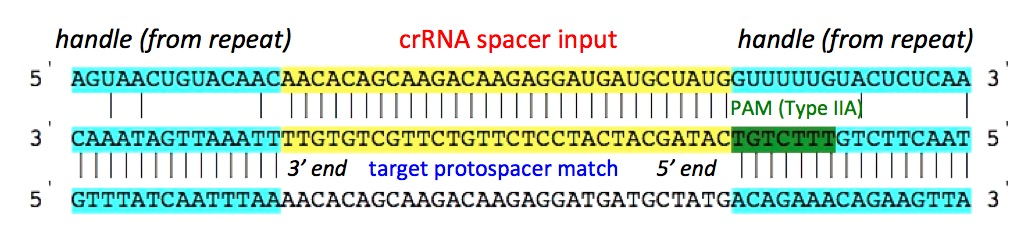
display. The spacer sequence is shown in the right side image.
|
   |
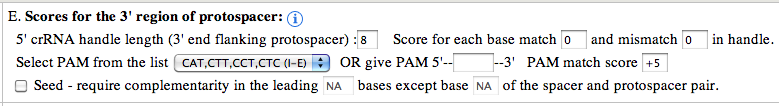
5' crRNA handle length : The default value used is 8, but user can increase/decrease the length of the handle ranging from 0 to any number (e.g: 100). There is no upper limit/restriction, but if the source sequence is not available, the length will be automatically adjusted. The handle sequence belongs to the repeat sequences (unless the handle length is greater than repeat sequence length). Score for each base match and mismatch : The default value set to 0, but user can alter the values to any positive or negative number (e.g: match: -1, mismatch: +1). If handle is present, these values can greatly help identifying the self matches. As, for self matches, the handle sequence of spacer will found all base pairing nucleotides. penalizing base pairing in the handle region will send the self matches down the order or even filter them out (using the cutoff score). Select PAM from the list : PAM (Protospacer Adjacent Motif) is often the key indicator of true positive CRISPR target match. It can be used to identify the targets of known CRISPR systems. The PAM types are shown below: I-A: NGG I-B: NGG I-E: CAT,CTT,CCT,CTC I-C: GAA I-F: GG Give PAM : User can also give a PAM motif (e.g: CGT). CRISPRTarget supports user given PAM to have IUPAC code. The following nucleotide codes are supported. IUPAC_code Base A Adenine C Cytosine G Guanine T/U Thymine (or Uracil) R A or G Y C or T S G or C W A or T K G or T M A or C B C or G or T D A or G or T H A or C or T V A or C or G N any base ./- gap PAM match score: The default value is +5, but can use any positive or negative integer value. PAM score can greatly help reordering the output and bring the true positive or target matches high in order. A combination of Spacer match/mismatch score, handle match/mismatch score and PAM match score along with cutoff score can greatly improve the outcome, specially when the output consists of several hundreds of matches. Seed require complementarity in the leading bases: This is one of the most important feature to directly filter out unsuitable matches from the output. As BLAST report may contain hits with partial match between spacer and protospacer. Often it doesn't start from the first base of the spacer, but for many CRISPR systems it's a must that the leading bases (adjacent to the PAM) should not have any mismatch, or a mismatch might be allowed (e.g: 5th base) but not to the other leading bases (e.g: no mismatch to the first 1 to 4, and 6 to 8). Researchers with such known models (properties), can apply the condition right at the very begining. For the said example, the input should be given as below: "Seed require complementarity in the leading 1-8 bases except base 5 of the spacer and protospacer pair." Note: if you want to exclude multiple bases, then give them comma separated. For the above example, if you want to exclude base 3 and 5, then give input as below: "Seed require complementarity in the leading 1-8 bases except base 3,5 of the spacer and protospacer pair."

3' crRNA handle length : The default value used is 8, but user can increase/decrease the length of the handle ranging from 0 to any number (e.g: 100). There is no upper limit/restriction, but if the source sequence is not available, the length will be automatically adjusted. The handle sequence belongs to the repeat sequences (unless the handle length is greater than repeat sequence length). Score for each base match and mismatch : The default value set to 0, but user can alter the values to any positive or negative number (e.g: match: -1, mismatch: +1). If handle is present, these values can greatly help identifying the self matches. As, for self matches, the handle sequence of spacer will found all base pairing nucleotides. penalizing base pairing in the handle region will send the self matches down the order or even filter them out (using the cutoff score). Select PAM from the list : PAM (Protospacer Adjacent Motif) is often the key indicator of true positive CRISPR target match. It can be used to identify the targets of known CRISPR systems. The PAM types are shown below: II-A: WTTCTNN,TTTYRNNN II-B: CNCCN,CCN Give PAM : User can also give a PAM motif (e.g: CGT). CRISPRTarget supports user given PAM to have IUPAC code. The following nucleotide codes are supported. IUPAC_code Base A Adenine C Cytosine G Guanine T/U Thymine (or Uracil) R A or G Y C or T S G or C W A or T K G or T M A or C B C or G or T D A or G or T H A or C or T V A or C or G N any base ./- gap PAM match score: The default value is +5, but can use any positive or negative integer value. PAM score can greatly help reordering the output and bring the true positive or target matches high in order. A combination of Spacer match/mismatch score, handle match/mismatch score and PAM match score along with cutoff score can greatly improve the outcome, specially when the output consists of several hundreds of matches. Seed require complementarity in the leading bases: This is one of the most important feature to directly filter out unsuitable matches from the output. As BLAST report may contain hits with partial match between spacer and protospacer. Often it doesn't start from the first base of the spacer, but for many CRISPR systems it's a must that the leading bases (adjacent to the PAM) should not have any mismatch, or a mismatch might be allowed (e.g: 5th base) but not to the other leading bases (e.g: no mismatch to the first 1 to 4, and 6 to 8). Researchers with such known models (properties), can apply the condition right at the very begining. For the said example, the input should be given as below: "Seed requires complementarity in the leading 1-8 bases except base 5 of the spacer and protospacer pair." Note: if you want to exclude multiple bases, then give them comma separated. For the above example, if you want to exclude base 3 and 5, then give input as below: "Seed requires complementarity in the leading 1-8 bases except base 3,5 of the spacer and protospacer pair."