17 7 2019. CRISPRDetect version 2.4 (download). https://github.com/davidchyou/CRISPRDetect_2.4
Version 2.4 has some bug fixes (compared to 2.2) , update to the CRISPRDirection (v2) module, and extends the CRISPR I-E identification of repeats. The array score is nowalso in the gff3 file. The gff3 file and txt files can be downloaded directly from the web output. The web interface uses a similar version with additions for the web inferface.
7 5 2018. Updated CRISPRDirection module with an increase in accuracy for Type II direction calls. Modified to handle gbff files (e.g. GCF or GCA) and cas gene annotations in these.
6 5 2016. CRISPRDetect is a tool to discover and explore the CRISPR non coding RNAs in sequence data. It is a bioinformatic tool to find CRISPR arrays. It is part of CRISPRSuite. The final submission version of CRISPRDetect v2.2 is described and available here. All earlier released presubmision '1' or 2.0 versions were testing versions and may have minor bugs.
News about CRISPRSuite is here:
For enquiries contact chris.brown at otago.ac.nz
1/6/2016 v2.1 The final submission version of CRISPRDetect v2.1 is described and available here. All earlier released presubmision '1' or 2.0 versions were testing versions and may have minor bugs, please update to 2.1 or later.
- If you use CRISPRDetect please cite:
Biswas, A., Staals, R.J. Morales, S.E. Fineran, P.C., and Brown,C.M. (2016) CRISPRDetect: A flexible algorithm to define CRISPR arrays BMC Genomics 17:365 link
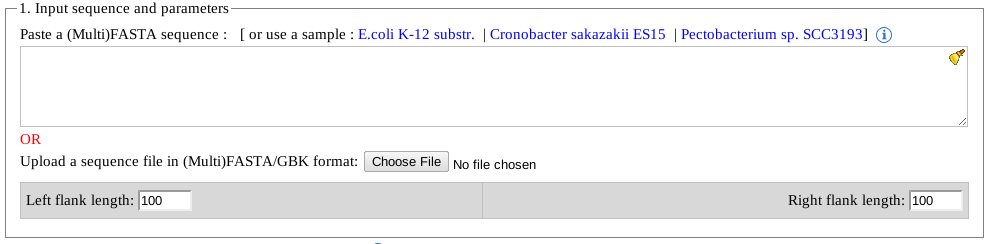
- FASTA/MultiFASTA sequences can be pasted in the text area.
- An NCBI accession with extension of .gbk or .gbfff can be given as input (e.g. NC_010473.gbk).
Note: Please note, in this format, multiple '.gbf' files are not supported (yet). Users are advised to input only one 'accession.gbk' at a time. Additionally, pasting the content of a '.gbk' file is not supported either. - Users can paste a direct link to the '.gbk' file from NCBI. For compatibility and security reasons, only NCBI links are supported at this time (e.g. ftp://ftp.ncbi.nlm.nih.gov/genomes/Bacteria/Pectobacterium_SCC3193_uid193707/NC_017845.gbk).
-
User(s) can paste the entire content of a NCBI annotation file (e.g. .gbk) in the text box including the genomic sequence at the bottom of the '.gbk' file.
Note: while downloading an annotation file (.gbk) from NCBI, or any other sources, make sure that the file is saved with genomic sequence and the downloaded file contains an extension of '.gbk'. -
CRISPRDetect output by default shows 100nt sequences upstream and downstream from all identified CRISPR arrays. These sequences can be useful for performing future analysis, as well as gives an idea if a CRISPR array may be extended or not.
Note: Please note, that unlike all the other optional parameters, the flank lengths can't be modified once an arrays is already predicted.

-
The term 'word' here refers to one of the two 11 nucleotide long (as default) genomic repeats separated by a non-identical genomic sequence stretch (potential spacer). CRISPRDetect allows any positive integer >7 as word length. However, based on our observation, most valid CRISPRs can be identified even with a higher (e.g. 13 or more) word length. Making the word length smaller will increase the number of putative CRISPRs, and processing time. Please note, that word length and minimum word repetition works together to find the putative CRISPRs. Increasing both might reduce identification of true CRISPRs, specially the shorter ones. (Valid range 7-n, default 11.)
-
Minimum word repetition works together with the word length to identify putative CRISPR. Increasing the number of repetitions drastically improves the speed as the list of false putative CRISPRs reduces, hence increases the processing speed to generate the output. However, while increasing the repetition, users should be careful not to increase both the word length and the number of repetition, as that may affect identification of true but shorter CRISPR arrays. (Vaild range 2-n, default 3.)
-
As the name suggests, the maximum gap between CRISPRs are used for joining two closely spaced CRISPRs with identical representative repeat. This parameter is also used as a cutoff for maximum spacer length. (Valid range any integer, default 125)

-
After the initial processing on the putative CRISPRs, the putative CRISPRs are checked to see if the minimum repeat lengths are >=11 (default) or a user provided positive integer >= word length, before further analysis is performed. The putative CRISPRs fails to meet this criteria are immediately discarded. (Valid range 7-n, default 11..)
-
After the initial processing on the putative CRISPRs, the putative CRISPRs are checked to see if they contains minimum 3 repeats (default) or a user provided positive integer >=2 before further analysis is performed. The putative CRISPRs fails to meet this criteria are immediately discarded. (Vaild range 2-n, default 3.)
-
The CRISPR likelihood score determines which CRISPRs to be shown in the final output. The CRISPRs with a score below user provided positive/negative number are moved to filtered out CRISPRs file. (Vaild range any integer, default 4.0.) Alternatives would be 0 or 0.5 to see all possible arrays, or 3.0 to see a few more possible arrays.

-
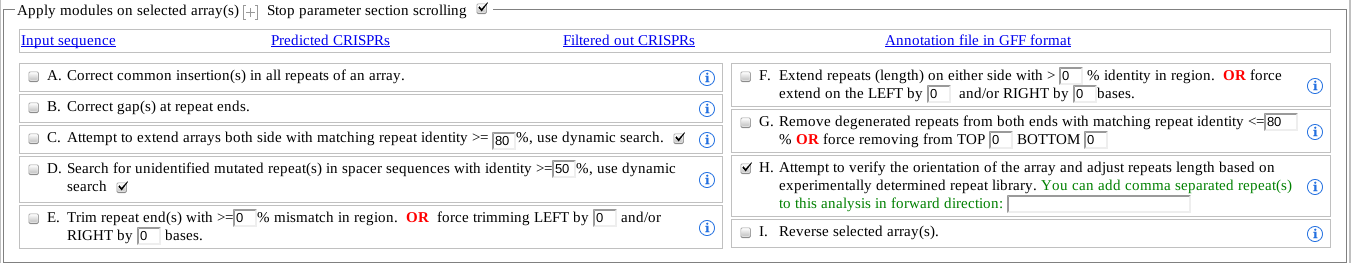
Common insertion(s) refers to gaps in the representative repeat of a CRISPR. This may occur when repeats are extended sidewise. This module is only applied when there are gap(s) in the representative repeat. Aids visualisation but does not change the data.
-
Repeats are extended on each side using an alignment of the repeats. This process may leave gaps symbols at the end of repeats. This module checks if a repeat has gaps at either end, and attempts to find the correct repeat bases from neighboring spacer sequences or flanks.
-
This module attempts to extend the CRISPRs from both ends with a matching repeat identity (default 55%). There is a lower limit of 35% identity as other wise the CRISPRs get extended rapidly with large degeneracy in the repeat.
The dynamic search is an intelligent way to determine if a degenerated repeat with identity (e.g. 50%) merginally below the identity cutoff (e.g. 55%) should be added to the CRISPR or not. When this option is selected, instead of comparing the repeat in question with the representative repeat, the closest repeat is used for the comparison. This option addresses the cases when one or more point mutation occurs at the end of CRISPrs and propagate to the next repeat(s) with added mutation. (Valid range 35-100%, defaul 55%.) -
As CRISPRs are extended in both ends with a default % identity (e.g. 67%), it may sometime fail to identify repeats with partial deletion at the ends, leaving the spacer exceptionally long. This module address this issue by searching for degenerated or repeat with multiple base deletions in all the spacers with length > representative repeat length and median spacer length. If dynamic search option is selected, then instead of using the representative repeat the previous repeat adjuscent to the spacer is used for comparison.
-
It is possible that automatic increase of the repeats ended up falsely predicting the boundaries, and the extra bases at the end should be removed using this module. When applied, the columns of the repeats are checked for mismatches. '0' means use defaul values.
-
CRISPRDetect attempt to extend the repeat length on both sides, comparing the bases from the following spacers and flanking regions with a dynamically obtained minimum percentage column identity. '0' means use defaul values.
-
Degenerated repeats can be falsely predicted by extending the CRISPRs and may require manual intervention to correct them, specially where the extension is true in one end and false in another. CRISPRDetect supports removal of degenerated repeats from either end, both automatically by using a minimum percentage similarity between the representative repeat and individual repeats, as well as by specifying the number of repeats and side of the array. In automatic shortening, the operation stops when a repeat shows identity above the cutoff or the minimum number of repeats (default 3) specified for initial array prediction. (Range 0-100%, default 66%.)
-
CRISPRDetect uses the algorithm described in CRISPRDirection algorithm. The CRISPRs identified to be wrongly oriented (i.e. the direction of transcription should be reverse), are automatically reversed. We strongly recomend users to use this module, as it has added functionality in correcting the repeat boundaries, and determining true positive CRISPRs. If the sequence of the repeat has been determined for your array enter it here. This will allow the program to get the direction and exact boundry correct (if it has not done so automatically)
-
This module reverses a CRISPR irrespective of the CRISPRs direction of transcription. However if the automatic direction finding module was also selected (H), higher priority is given to the automatic direction finding module- so unselect H.
-
Although, most of these modules are part of the default workflow, often some CRISPRs require further processing and use of funtionalities in different order to represent the CRISPR more accurately. The modules numbered with A-I, allows users the flexibility to perform additional analysis in an interactive way, with abilities to Undo/Redo any action performed. Through the following example we will explain this in detail.
Example 1: Lets assume a predicted CRISPR has a mutated or highly degenerated repeat at the 3' end. The amount of degeneracy is slightly less that the default. Now, by using a lower repeat identity (say 45%), the user can extend the CRISPR to include the missing repeat. However, while doing this, a non-repeat (piece of sequence with multiple gaps and mismatches, but with identity of 45%) may also get included in the 5' end.
User now has the following choices :
a). Click the Undo button and increase the identity to omit the wrongly predicted repeat.
b). Use module G (i.e. Remove degenerated repeats) and specify number of repeats (i.e. 1) to be removed from the intended end (i.e. TOP). Example 2: The direction of the CRISPR transcription is analysed using CRISPRDirection algorithm. One major component in CRISPRDirection algorithm is to compare the direct repeat against experimentally verified repeat library. However, it is possible, that a user may have his/her own verified repeat which are not part of the repeat library (yet). The direction of all the CRISPRs that contains this experimentally verified repeat can be checked or fixed by simply inputting the repeat sequence in 5'-3' order in the given text box in option H.

-
User can select one or more modules from the A-I options, and apply them on one or more CRISPR arrays. There are built in functions to select All, All questionable and All unquestionable arrays by changing the option from the drop-down list. Alternately, user can manually select CRISPRs and re-run the intended analysis.
-
Multiple edits like force removal of repeats from ends or trimming of repeats can change a CRISPR to a largely different CRISPR, and a restoration of the CRISPR might be required. This can be done by clicking the 'Restore arrays' button. This button will restore all arrays to their original form irrespective of their selection. If, only selected arrays need to be restored, then user need to use the 'Undo' buttons in each of those arrays.
-
This option extract the representative repeat from all the selected arrays, and give them in FASTA format.
Example:
>NC_017845|Pectobacterium-sp. SCC3193, complete genome.
GTGTTCCCCGCGCCAGCGGGGATAAACCG -
This option will extract the spacer sequences from all the selected arrays, and give them in FASTA format as shown below. The options, 'All' and 'Good' referes to the quality of the spacers. The 'All' options extracts all the spacers irrespective of the presence of partial/full deletion in the spacers, where 'Good' spacers only include spacers without any deletion.
Note: Whether a spacer has undergone deletion is determined by the length of the spacer, compared against the 'median' length of all the spacers in an CRISPR array and not applicable for short CRISPRs with less than 3 spacers. A spacer will be termed 'Good' when its length is >=80% of the 'median' length.
Example:
>NC_017845|Pectobacterium-sp. SCC3193, complete genome.|2_451274_452587|2_1_451303_451334|32, Direction:Forward TCCAATTTAAATATGCTTTTTTTACCAATTCC >NC_017845|Pectobacterium-sp. SCC3193, complete genome.|2_451274_452587|2_2_451364_451396|33, Direction:Forward GACGAGGAATCTGATAACGCATTCCGTGTTCGC >NC_017845|Pectobacterium-sp. SCC3193, complete genome.|2_451274_452587|2_3_451426_451457|32, Direction:Forward GTCGTTCATCAGGTCATGCTCTGACAGCTTCG >NC_017845|Pectobacterium-sp. SCC3193, complete genome.|2_451274_452587|2_4_451487_451518|32, Direction:Forward CAGACCCTCTTTGGTATACTTCAATTTTATAA >NC_017845|Pectobacterium-sp. SCC3193, complete genome.|2_451274_452587|2_5_451548_451579|32, Direction:Forward CAACGACATCGAAATCAGCGCTAAATATTTCT >NC_017845|Pectobacterium-sp. SCC3193, complete genome.|2_451274_452587|2_6_451609_451640|32, Direction:Forward GATGCGATGAAACACACGTATCTGGGCTATGC >NC_017845|Pectobacterium-sp. SCC3193, complete genome.|2_451274_452587|2_7_451670_451702|33, Direction:Forward TGGAAGGCTGGGCGCGTGAAATCGGGTGCAATC >NC_017845|Pectobacterium-sp. SCC3193, complete genome.|2_451274_452587|2_8_451732_451763|32, Direction:Forward TCTGTCATTGGCTGTCGGGGAGGTTCACTGCC >NC_017845|Pectobacterium-sp. SCC3193, complete genome.|2_451274_452587|2_9_451793_451824|32, Direction:Forward CCCCAGCGCATAAACGACGCTATACCACAACC >NC_017845|Pectobacterium-sp. SCC3193, complete genome.|2_451274_452587|2_10_451854_451885|32, Direction:Forward TCAGCCCCCCAAACAAACACAGCCGCTATCCA >NC_017845|Pectobacterium-sp. SCC3193, complete genome.|2_451274_452587|2_11_451915_451946|32, Direction:Forward TCTGCGTTAACAGGCCGCTTGTTTGGGCGTTT >NC_017845|Pectobacterium-sp. SCC3193, complete genome.|2_451274_452587|2_12_451976_452007|32, Direction:Forward ATAAATCATATTCAGAGTTGGAATTCTTGAAT >NC_017845|Pectobacterium-sp. SCC3193, complete genome.|2_451274_452587|2_13_452037_452068|32, Direction:Forward CTGTGGCTGAGCTATTTCGTGCGCGTCGTTGA >NC_017845|Pectobacterium-sp. SCC3193, complete genome.|2_451274_452587|2_14_452098_452129|32, Direction:Forward AGAAAAATACTCAATCGTGGTCGTGCCGAAAC >NC_017845|Pectobacterium-sp. SCC3193, complete genome.|2_451274_452587|2_15_452159_452190|32, Direction:Forward CTGTTTGGGTTGATGAGAAGGAGAGGACTTTG >NC_017845|Pectobacterium-sp. SCC3193, complete genome.|2_451274_452587|2_16_452220_452251|32, Direction:Forward GATATGCAAAACGTCATTATCGGCGGCATTCT >NC_017845|Pectobacterium-sp. SCC3193, complete genome.|2_451274_452587|2_17_452281_452312|32, Direction:Forward GTTGCCGAAATCTTCAATGGCATGGATTTAGG >NC_017845|Pectobacterium-sp. SCC3193, complete genome.|2_451274_452587|2_18_452342_452373|32, Direction:Forward AGAAAAATACTCAATCGTGGTCGTGCCGAAAC >NC_017845|Pectobacterium-sp. SCC3193, complete genome.|2_451274_452587|2_19_452403_452434|32, Direction:Forward TGCTCGCAACGTTTGCGTATCAGGACTACGCA >NC_017845|Pectobacterium-sp. SCC3193, complete genome.|2_451274_452587|2_20_452464_452496|33, Direction:Forward TGCTAATTTGTAACCTTCCGGTATTACCGGAGA >NC_017845|Pectobacterium-sp. SCC3193, complete genome.|2_451274_452587|2_21_452526_452557|32, Direction:Forward TTCCAACCCTTTCAGCAAGCTCTACCTGAGTA
Description: CRISPRDetect preserves all the positional information for each spacer. It contains both the start and stop position as well as the index of the spacers. For example, '2_1_451303_451334|32' indicates, that the spacer is the 1st spacer (from top or 5' end) of the 1st CRISPR from Pectobacterium-sp. SCC3193, and the spacer is 32nt long and in forward direction (i.e. 5' to 3').
Note: Please remember to allow "pop-up" windows for "http://bioanalysis.otago.ac.nz/" or this function may not be available.
-
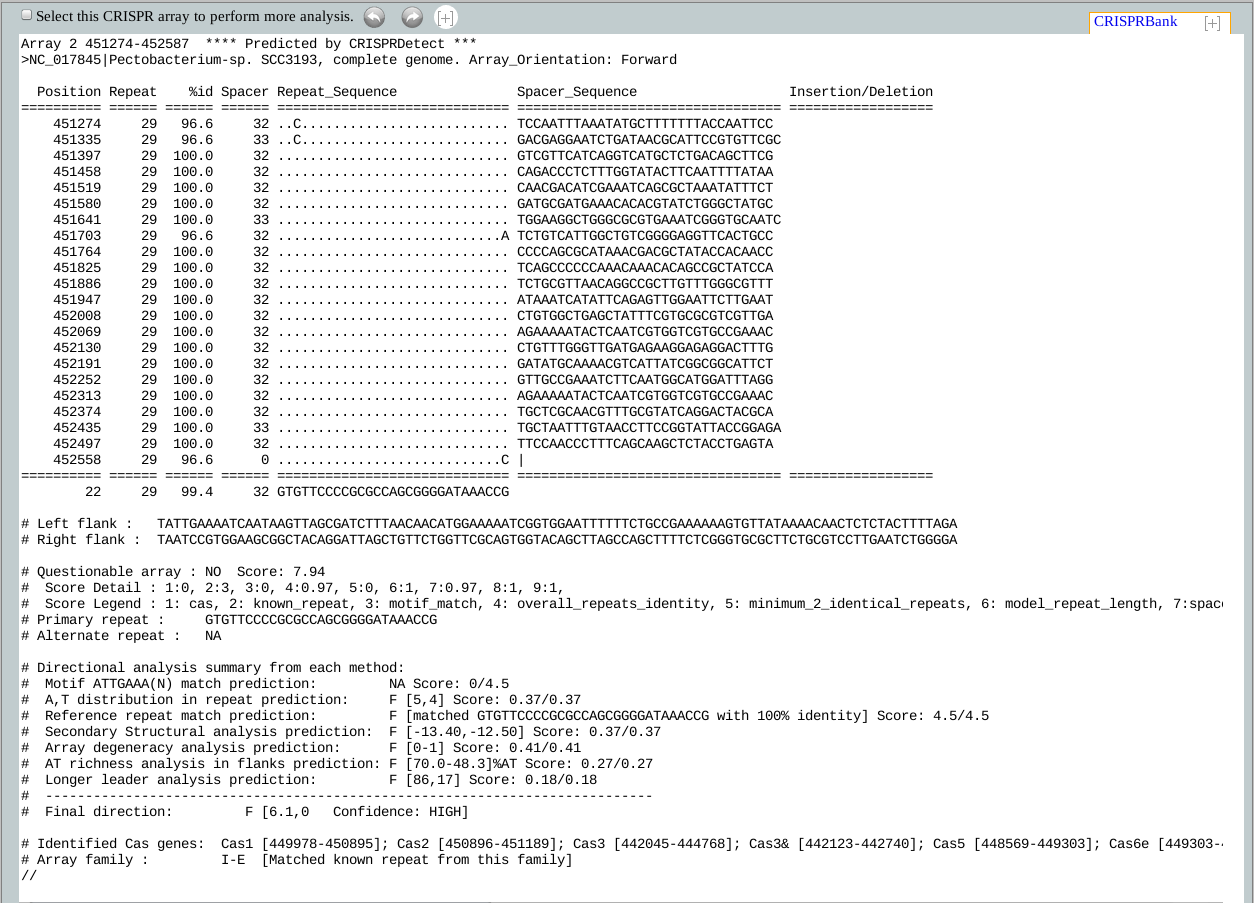
The top most line of the CRISPR arrays contain the positional information followed by "**** Predicted by CRISPRDetect 2.1 ***" which is important to be retained if the array will be further processed with other tools from our lab, such as CRISPRTarget. CRISPRTarget look for this line to identify which program predicted the array and extracts information accordingly.
-
The 2nd line from the top contains the source of the CRISPR.
-
Column headers: The 4th line from the top shows the column headers of the array and explained below:
Position: Position refers to the start position of the current repeat. For arrays in forward direction
Repeat: This field shows the repeat length.
%id: This field refers to % identity of the current repeat against the representative repeat.
Spacer: This field shows the current spacer length.
Repeat_Sequence: As the term suggests, this field shows the alignment of the current repeat with the representative repeat. The dots means indentical base, '-' means gap, and A/T/G/C refers to mismatch.
Spacer_Sequence: This column shows the current spacer sequence.
Insertion/Deletion: Insertion(s) and deletions are very common to CRISPRs and well supported by CRISPRDetect.
Insertion(s):Instead of showing the inserted bases in the repeat, which will affect the repeats alignment against the representative repeat, CRISPRDetect move the bases to this coulmn and shows their position in the genome. In case of operations like array shortening or repeat length extension/shortening, CRISPRDetect always checks if there are any inserted bases which needed to be considered.
Deletions(s):For deletion in direct repeats, some existing tools takes equal number of bases from the adjuscent spacers to make the repeats of equal length, making both the repeat and spacer incorrect. In CRISPRDetect, when a repeat/spacer loses one or more bases through deletion, the lost bases from the repeats are replaced with '-'s and a relative deletion notification is shown in this column with the starting position. For spacers, when a spacer found to have length < 80% of the median length of all the spacers from an array (applicable to arrays with 3 or more spacers), it is considered to be a 'spacer with deletion'. These cases are notified in this column with relative position(s).
-
The 2nd line after the last repeat (i.e. the last spacer is denoted with '|') contains total number of repeats (i.e. 22 in the above example), average repeat length, average % identity of all repeats, average spacer length and representative repeat of the array.
-
Left flank: This line contains a short piece of sequence that can be referred as leader sequence (sequence from the 5' end of a CRISPR).
-
Right flank: This line contains a short piece of sequence that can be referred as trailer sequence (sequence from the 3' end of a CRISPR).
-
Questionable array: CRISPRDetect uses a sophisticated scoring method to determine the quality of the CRISPRs. The detail of the scoring method can be found in the supplement to the paper.. Based on this array quality score, a CRISPR can be categorized as either 'Questionable' or poor array (when the score is < 4.0), or 'Unquestionable' or good array (when the score is >=4.0).
-
Subscores-
1. Presence of either cas1 or cas2 genes in the genome is awarded ( +1, or 0). (cas)
This method is only applied when an annotation file (NCBI gbk or gbff file) is used as input. The annotation files are searched (term based) to create a list of all cas genes present in the genome. The scoring system awards the quality score with ‘+1’ when annotation of either cas1 or cas2 genes are present in the input file.
2. Match to known repeat using a set of reference repeats from high confidence arrays (+3). (likely repeat)
We use 26 experimentally verified representative repeats as reference and increased the set of known repeats by allowing up to 7 base mismatches. This extended set of ~400 repeat was used to predict a higher confidence set. Arrays were predicted then those with greater than 7 repeats and scores > 4 were used to predict a set of likely repeats. This file was converted in to a BLAST database and potential repeat searched against that with blastn-short which is optimised for short sequences. When a match is found, the array quality score is awarded ‘+3’. This file or the score can be modified in the commandline version.3. Repeat has at least 23 bases and ATTGAAA(N) at the end (+3, or 0). (motif_match)
Another feature adapted from the CRISPRDirection algorithm is the presence of motif ATTGAAA(N) at the 3’ end of repeats. We observed that, this motif is an accurate indicator of the direction of transcription. In that paper we also observed that all the potential repeats that are >=23nt long containing this motif were genuine CRISPRs. Hence, we used this information to contribute to the quality score, and the quality score is awarded with ‘+3’ when the repeats are >=23nt long and contains ATTGAAA(N) at the 3' end.4. Overall repeat identity within an array (0 to 1). (overall_repeat_identity)
The overall repeats identity score (S) is calculated using the following method
S= (average % identity of the repeats - 80)/20
The maximum possible positive score can be 1 (when all repeats are identical). However, the score will be negative, when the overall repeat identity is <80%.5. The repeats in the array do not form one sequence similarity cluster (-1.5, or 0). (one_repeat_cluster)
The repeat are clustered using CD-HIT-EST if they form more than one cluster the quality score is penalized by ‘-1.5’.6. Scoring the repeat lengths (range -3 to +1). (exp_repeat_length)
In this method, we use the table of repeat length distribution (Figure 3). The relative score (S) for a repeat of length (L) is determined using the following rules:
S= 0.25 + L/H [where, L>=23 and L =< 47;
H is the most abundant repeat length for bacteria or archaea]
S= -0.25*(23 - L) [where, L <23]
S= -0.25*(L - 47) [where, L >47]
The maximum negative score limit is set to -3, and maximum positive score limit is +1.7. Scoring the spacer lengths (range -3 to +3). (exp_spacer_length)
In this method, each spacer of an array is independently scored, and counted towards a final spacer length score. The individual spacer length score (S) for a spacer with length (L) within the range 28-48 (see Fig 3B) are awarded a positive score using the formula:
S = 0.01 + N/H [where, 27< L =<48;
N= Total number of spacers of this length;
H= Most abundant spacer length for bacteria or archaeaAny spacer length outside this range is penalised by the following rule:
S=-0.10* (28 - L) [where, L<28]
S=-0.10* (L -48) [where, L>48]Finally, an average spacer score for the current array is calculated using
Average score=Sum_of_scores/no_of_spacersThe maximum negative score limit is -3 and maximum positive score limit is +1.
8. Overall spacer identity (-3 to +1) (spacer_identity)
In this method we test the sequence (dis)similarity among all the spacers. If the spacers are all near identical it is more likely to be a direct repeat, possibly a tandem repeat rather than a CRISPR array. If the spacers belong to a total number of clusters (C) with identity >=80%, the spacer identity score (S) for an array with number of spacers (N) is calculated using the following rule:
S= -3 [where, C =< integer (N/2); ]
S= 0.20*C [where, C > integer (N/2); ]
The positive score limit is +1.9. Scoring total number of identical repeats 0 to +1) (log(total repeats) - log(total mutated repeats))
Since longer arrays, and those with a greater number of identical repeats are more likely to be a true CRISPR, this scoring method uses both. If an array contains ‘P’ identical repeats out of the ‘N’ total number of repeats, then the score (S) is calculated using the following rule:S= log (N) - log (N-P) [where, P=Identical repeats, N= total number of repeats]
The maximum positive score limit is +1.